



Structural comparison and chemical analysis of various types of anodic aluminum oxide (AAO) finishes produced from conventional electrolytes and those produced from an electrolyte modified with electroactive polymer determined differences, which explain superior bonding performance by the AAO produced from the modified electrolyte.
An extensive study comparing the adhesion of various inks and adhesives to the modified oxide and to Types I, II and III anodic oxide finishes elucidated the reaction and bonding mechanism of the supplementary coatings to the AAO. By understanding this mechanism, a basis for organic-inorganic reactions within anodic oxides can be realized, resulting in bonds with higher reliability, offering new areas for AAO application.
Introduction
 Dr. Jude RungeAdhesive joints can be utilized to bond any variety of materials. With lightweighting in the automotive industry increasing the usage of aluminum, the demand for adhesive bonding between components is on the increase.1 This paper deals specifically with joints that involve aluminum components. Regardless of the bonding partners, the fundamental adhesive attachment to metal is a polymer-metal bond.
Dr. Jude RungeAdhesive joints can be utilized to bond any variety of materials. With lightweighting in the automotive industry increasing the usage of aluminum, the demand for adhesive bonding between components is on the increase.1 This paper deals specifically with joints that involve aluminum components. Regardless of the bonding partners, the fundamental adhesive attachment to metal is a polymer-metal bond.
Adhesive joints can be enhanced by way of a variety of methods including: mechanical and/or chemical roughening; and chemical adhesion promoters, such as conversion coatings, and other types of primers. Anodic oxides are also utilized to enable adhesion of a polymer layer to a metal substrate, be it an ink, paint, lacquer or adhesive. The key to a reliable bond is often the transition layer between the aluminum and subsequent organic adhesive. The purpose of this paper is to present the theory and mechanism for enhancing the anodic aluminum oxide by way of the addition of electroactive polymer to the standard electrolyte. Examples of how the finish, modified with electroactive polymer, is applied as a transition layer for bonding purposes today are also presented.
Aluminum Surface Reactions
The passive layer
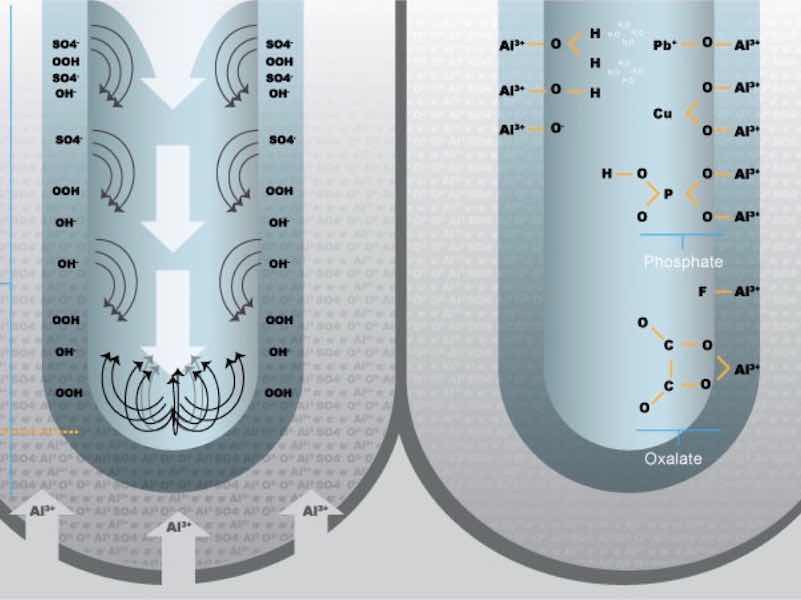
Under atmospheric conditions, surface reconstruction, by way of oxidation, proceeds extremely rapidly on clean aluminum, as a stable passive film of the thickness of several molecular layers is formed. The natural oxide layer always contains a certain amount of aluminum oxihydrate (AlOOH) and water bonded physically or by chemisorption. The initial reaction is preferential, at energetically favorable reaction sites, with chemisorption first occuring at certain surface structures, crytallographic orientations and other defects. For the oxide layer to grow, diffusion of either the oxygen from the atmosphere or the metal through the oxide layer or both, is required. The nuclei mature (ripen) to form stable oxide nodes that grow due to the chemical potential difference between the oxide and the base metal. The larger nodes grow laterally and perpendicularly at the expense of the smaller nodes to form a layer. The resultant layer is very thin and sensitive to the environment in which it forms.2,3 A final passive layer thickness of approximately 10 nanometers can be reached after a prolonged exposure time. See Figure 1.
 Figure 1: Passive layer growth schematic
Figure 1: Passive layer growth schematic
Sato4 classifies the passive oxide films into two categories: (1) the network former; and (2) the network modifier. Aluminum falls into the category of a network former, meaning it forms a layer comprised of a single oxide type (Al2O3). The anodic formation of network-forming oxides most likely occurs due to the inward oxide ion migration to the metal-oxide interface and produces a compact layer containing no foreign anions other than oxide ions. In contrast, passive layers on metals, other than aluminum, are classified as network modifiers, forming a multilayered oxide film of progressive stoichiometry, such as cobalt oxide, which forms an inner divalent oxide layer and an outer trivalent oxide layer (Co/CoO/Co2O3). Network-modifying oxide growth is attributed to outward metal ion migration to the oxide-electrolyte interface, which forms, unlike the passive film on aluminum, a defective film that contains foreign anions.
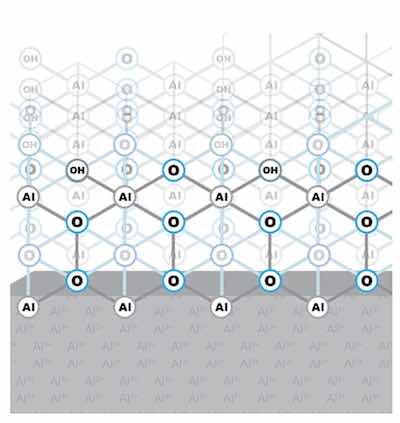 Figure 2: Passive layer chemical structureThe concept of the formation of a progressive networking-forming oxide layer on aluminum and aluminum alloys is important to understanding some key characteristics and properties of the anodic oxide, and how the anodic oxide nucleates and grows. The structure of the passive film is characterized as amorphous. Nevertheless, a layer develops that has a chemical structure. This structure provides the path for ionic conduction and continued oxide growth. As the layer structure develops, at the aluminum-oxide interface, no ionic tunneling is allowed to occur across the passive layer because the oxide network constitutes a barrier layer to ion transfer from the outside, but does not inhibit outward electron transfer, which maintains the exclusive aluminum-oxygen constitutive make-up of the passive layer. See Figure 2.
Figure 2: Passive layer chemical structureThe concept of the formation of a progressive networking-forming oxide layer on aluminum and aluminum alloys is important to understanding some key characteristics and properties of the anodic oxide, and how the anodic oxide nucleates and grows. The structure of the passive film is characterized as amorphous. Nevertheless, a layer develops that has a chemical structure. This structure provides the path for ionic conduction and continued oxide growth. As the layer structure develops, at the aluminum-oxide interface, no ionic tunneling is allowed to occur across the passive layer because the oxide network constitutes a barrier layer to ion transfer from the outside, but does not inhibit outward electron transfer, which maintains the exclusive aluminum-oxygen constitutive make-up of the passive layer. See Figure 2.
Once a stable passive oxide forms, it may be transformed by hydration to hydroxide or hydrate compounds in chain-like coupling, forming a reactive structure between the atmosphere and the base metal. However, because the passive film is extremely thin, on the order of 1 to 10 nanometers, it is fragile and easily subject to abrasion. Nevertheless, the passive layer provides corrosion resistance to the underlying metal, until it is scratched or otherwise disturbed. If the atmospheric conditions do not change, the layer heals itself by reforming.
Conversion coatings
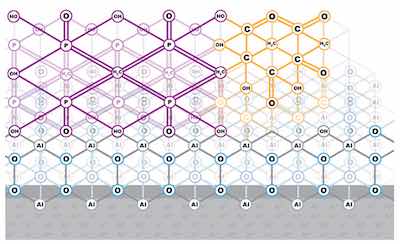 Figure 3: Schematic of passive layer enhanced by the adsorption of vinyl phosphonic acid and acrylic acid, a common conversion layer used as a primer layer for bonding polymers to aluminum.The barrier layer phenomenon of the network-forming passive layer is the basis for the reactions that can be employed to enhance the efficacy of the naturally occurring oxide in a variety of applications of the aluminum substrate, including the function as a transition layer. Because the oxide will not allow the penetration of external anions, the anions can adsorb at active sites on the outer chemical structure of the oxide. “Conversion” of the structure to other compounds by the adsorption of a supplementary anion imparts different or additional properties to the passive layer at the surface. This is an equilibrium process, with no external driving force such as an electrical power supply.
Figure 3: Schematic of passive layer enhanced by the adsorption of vinyl phosphonic acid and acrylic acid, a common conversion layer used as a primer layer for bonding polymers to aluminum.The barrier layer phenomenon of the network-forming passive layer is the basis for the reactions that can be employed to enhance the efficacy of the naturally occurring oxide in a variety of applications of the aluminum substrate, including the function as a transition layer. Because the oxide will not allow the penetration of external anions, the anions can adsorb at active sites on the outer chemical structure of the oxide. “Conversion” of the structure to other compounds by the adsorption of a supplementary anion imparts different or additional properties to the passive layer at the surface. This is an equilibrium process, with no external driving force such as an electrical power supply.
Formulations for conversion coatings that contain chromates, phosphates and silicates, to name just a few, are utilized to bolster corrosion resistance and wear resistance, as well as to serve as a primer layer for paints or adhesives. As is, the finishes are rarely thicker than the 1 to 10 nanometers, which comprise the unmodified passive layer. Other polymer modifiers can be introduced, which increase the conversion-coating layer thickness by virtue of long hydrocarbon chain length. See Figures 3 and 4.
 Figure 4: Transmission Electron Microscope(ic) (TEM) image of a vinyl phosphonic acid conversion- coated aluminum alloy enhanced with a polymer modifier (acrylic acid). The initial thickness of the conversion coating, without the modifier, was reported to be the same thickness as the passive layer on the aluminum (maximum 10 nanometers). By adding acrylic acid as a long-chain modifier to the conversion coating formulation, additional thickness was obtained. The final layer measured 15 nanometers thick.The number of sites on the passive oxide structure, on which supplementary anions can adsorb, limits the reaction and film-building attributes of a conversion coating. The oxidation reaction, driven by the presence of available aluminum, slows down and eventually stops when the aluminum at the interface has been completely reacted. Therefore, conversion coatings will always be very thin. Nevertheless, conversion coatings are often used as a transition layer since they are chemically bound to the aluminum surface by way of the naturally occurring passive oxide. Supplementary anions, bound to the surface of the passive layer structure, provide the vehicle to form the transition bond.
Figure 4: Transmission Electron Microscope(ic) (TEM) image of a vinyl phosphonic acid conversion- coated aluminum alloy enhanced with a polymer modifier (acrylic acid). The initial thickness of the conversion coating, without the modifier, was reported to be the same thickness as the passive layer on the aluminum (maximum 10 nanometers). By adding acrylic acid as a long-chain modifier to the conversion coating formulation, additional thickness was obtained. The final layer measured 15 nanometers thick.The number of sites on the passive oxide structure, on which supplementary anions can adsorb, limits the reaction and film-building attributes of a conversion coating. The oxidation reaction, driven by the presence of available aluminum, slows down and eventually stops when the aluminum at the interface has been completely reacted. Therefore, conversion coatings will always be very thin. Nevertheless, conversion coatings are often used as a transition layer since they are chemically bound to the aluminum surface by way of the naturally occurring passive oxide. Supplementary anions, bound to the surface of the passive layer structure, provide the vehicle to form the transition bond.
Anodic oxides
When the nucleation and growth of the passive layer is enhanced by stimulating the electrochemical process of its formation with an external current, the result is the growth of an anodic oxide. By polarizing the surface and flooding the interface with aluminum ions, an ordered oxide structure grows from the surface. Just like the passive layer, the AAO functions as a barrier layer to the intrusion of external ions from the electrolyte. Therefore, anions adsorb on the surface of the AAO that comes in contact with the electrolyte.
It is well documented that by changing the anodizing process parameters, the properties of the resultant finish change. This is so well established that the metal finishing industry recognizes three different basic anodizing finishes: Type I (chromic acid anodizing); Type II (sulfuric acid anodizing); and Type III (sulfuric acid hard anodizing). Each has different engineering properties and, correspondingly, different microstructures. From the appearance of standard finish microstructures, it is easy to see that the anodizing process parameters of current density, temperature and formulation impact the structure and the engineering properties and finish performance.5
 Figure 5: TEM image of a Type I or chromic acid anodic oxide. AAO produced in chromic acid doesn’t show a columnar structure, but it is spongy and porous. The random structure prohibits finish growth beyond 3 to 5 microns.The irregular, skewed oxide structure of the Type I finish explains why at even thin finish thicknesses (1 to 3 microns), the chromic acid finish exhibits good corrosion resistance. Without distinct unidirectional pores, environmental ingress is reduced significantly. Further processing the Type I finish with inert seals, or using it as a primer, enhance the corrosion resistance of the finished component even more. Its naturally thin finish thickness is ideal for applications that demand high-tolerance control and fatigue performance. See Figure 5.
Figure 5: TEM image of a Type I or chromic acid anodic oxide. AAO produced in chromic acid doesn’t show a columnar structure, but it is spongy and porous. The random structure prohibits finish growth beyond 3 to 5 microns.The irregular, skewed oxide structure of the Type I finish explains why at even thin finish thicknesses (1 to 3 microns), the chromic acid finish exhibits good corrosion resistance. Without distinct unidirectional pores, environmental ingress is reduced significantly. Further processing the Type I finish with inert seals, or using it as a primer, enhance the corrosion resistance of the finished component even more. Its naturally thin finish thickness is ideal for applications that demand high-tolerance control and fatigue performance. See Figure 5.
The fine, highly ordered, unidirectional, nanoscale porous columnar microstructure of the Type II finish explains why it can be utilized for decorative applications. At finish thicknesses ranging from 5 to 20 microns, the sulfuric acid finish exhibits a high, fine-pore density and brings little change to the color of the substrate. Hence, the name “Clear Coat.” Type II anodizing has a niche in the metal-finishing industry that is synonymous with color finishes for aluminum. As anodized, the porous structure of the anodic oxide is polar, which lends itself to bonding with supplementary coatings that may be inorganic or organic. A technique called “flash anodizing,” based on the process for Type II anodizing, yields thin AAO finishes that measure only several nanometers thick. The AAO produced by way of flash anodizing develops a uniform and continuous porous oxide from sulfuric, oxalic or phosphoric acid electrolytes, and is used as a stable and robust interface to enhance bonding. Because it is an integral oxide, it eliminates the concern for filiform corrosion. See Figure 6.
 Figure 6: (Left) TEM image of a Type II or “clear coat” anodic oxide on 6061 T6 aluminum. (Right) TEM image of a “flash anodized” 6061 T6 aluminum.The thick, heavy, highly ordered, unidirectional nanoscale porous columnar microstructure of the Type III finish, or hard anodic oxide, explains why it is a wear- and corrosion-resistant workhorse in the metal-finishing industry. In applications where the component is subject to impact, or has other mating wear surfaces, bonding can be a requirement for a Type III finish. See Figure 7.
Figure 6: (Left) TEM image of a Type II or “clear coat” anodic oxide on 6061 T6 aluminum. (Right) TEM image of a “flash anodized” 6061 T6 aluminum.The thick, heavy, highly ordered, unidirectional nanoscale porous columnar microstructure of the Type III finish, or hard anodic oxide, explains why it is a wear- and corrosion-resistant workhorse in the metal-finishing industry. In applications where the component is subject to impact, or has other mating wear surfaces, bonding can be a requirement for a Type III finish. See Figure 7.
The clear differences in anodic oxide microstructure, that directly depend on process parameters, infer that other process changes will also induce microstructural changes. By studying finishes anodized following standard processes and comparing those finished with different process parameters, a greater understanding of the mechanism of porous oxide formation has been obtained. These studies emphasize the need to understand the reactions that occur in the finish as it forms and the function of the various parts of the finish.
Electrolyte modification and anodic oxide structure
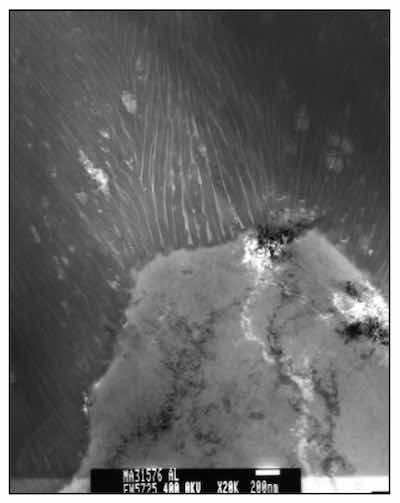 Figure 7: TEM image of a Type III or hard anodic oxide. The thicker column walls and proportionally wider pores offer more wear resistance when compared to Type II or Type I anodic oxides.For the purpose of this paper, attention will focus on modifications to the electrolyte or chemical modifications to the anodizing process. Our studies have shown that when considering the ramifications of a chemical change, the impact that the change will have on the central pore of the anodic oxide finish is of utmost importance. This is because the electrolyte contacts the porous structure, while the anion, which is in part oxygen, reacts with aluminum ions diffusing through the finish at the pore surface. Anion species (OH-, SO3-, CrO3-, etc.) are adsorbed on the pore walls, facilitating conduction and maintaining the net negative charge of the anodic oxide and the erect nature of each column. However, these adsorbed anions also provide individual reaction sites for the growing finish. Therefore, modifier selection depends upon finding a binding molecule for these reaction sites. Modifiers can chemically reduce these sites to the point of total column dissolution, as in sealing, or oxidize the sites to the point of completely shutting down ion exchange in the pore, as does the chromate ion in a Type I anodic oxide finish. Unmodified, the porous structure of all AAO is hygroscopic (absorbs atmospheric water), which is the principle for sealing of the anodic oxide. This attribute limits the time over which a bond external to the AAO can be made. See Figure 8.
Figure 7: TEM image of a Type III or hard anodic oxide. The thicker column walls and proportionally wider pores offer more wear resistance when compared to Type II or Type I anodic oxides.For the purpose of this paper, attention will focus on modifications to the electrolyte or chemical modifications to the anodizing process. Our studies have shown that when considering the ramifications of a chemical change, the impact that the change will have on the central pore of the anodic oxide finish is of utmost importance. This is because the electrolyte contacts the porous structure, while the anion, which is in part oxygen, reacts with aluminum ions diffusing through the finish at the pore surface. Anion species (OH-, SO3-, CrO3-, etc.) are adsorbed on the pore walls, facilitating conduction and maintaining the net negative charge of the anodic oxide and the erect nature of each column. However, these adsorbed anions also provide individual reaction sites for the growing finish. Therefore, modifier selection depends upon finding a binding molecule for these reaction sites. Modifiers can chemically reduce these sites to the point of total column dissolution, as in sealing, or oxidize the sites to the point of completely shutting down ion exchange in the pore, as does the chromate ion in a Type I anodic oxide finish. Unmodified, the porous structure of all AAO is hygroscopic (absorbs atmospheric water), which is the principle for sealing of the anodic oxide. This attribute limits the time over which a bond external to the AAO can be made. See Figure 8.
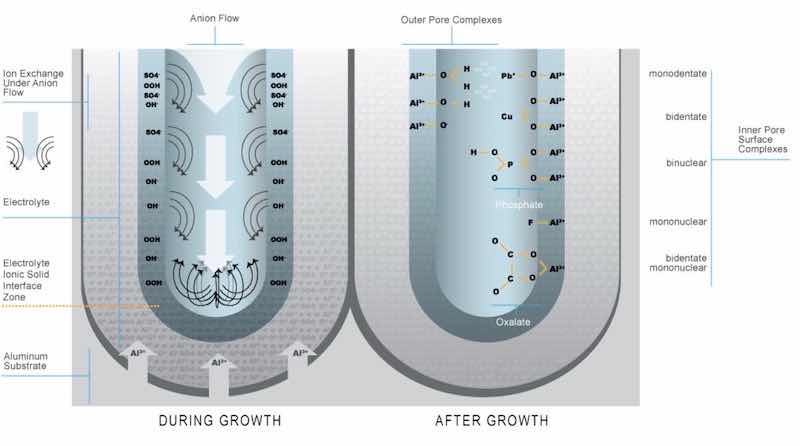 Figure 8A: As anodizing proceeds, the anion can be adsorbed on the surfaces exposed to the electrolyte.6 Figure 8B: The charged surface of the column wall provides adsorption sites for reactive species in the electrolyte such as modifiers and contaminants. These sites are also the critical adsorption sites for seals and other electroactive post-anodizing process treatments.7Understanding the nature of the central pore, what comprises the column walls and how they function are key to understanding how and what type of electrolyte modification will effect change of the anodic oxide structure. Electroactive modifiers – those that achieve and maintain conductivity within the columnar structure through introduction to the acid electrolyte – will generally participate in the anodizing reaction, and may be included within the finish as it grows. Insulating modifiers do not otherwise participate in the anodic oxide finish formation. However, glycerin-based electrolyte additives may act as surface-active agents or buffers that reduce the heat of reaction at the oxide-aluminum interface by way of saponification in order to promote a smoother, more uniform finish thickness.
Figure 8A: As anodizing proceeds, the anion can be adsorbed on the surfaces exposed to the electrolyte.6 Figure 8B: The charged surface of the column wall provides adsorption sites for reactive species in the electrolyte such as modifiers and contaminants. These sites are also the critical adsorption sites for seals and other electroactive post-anodizing process treatments.7Understanding the nature of the central pore, what comprises the column walls and how they function are key to understanding how and what type of electrolyte modification will effect change of the anodic oxide structure. Electroactive modifiers – those that achieve and maintain conductivity within the columnar structure through introduction to the acid electrolyte – will generally participate in the anodizing reaction, and may be included within the finish as it grows. Insulating modifiers do not otherwise participate in the anodic oxide finish formation. However, glycerin-based electrolyte additives may act as surface-active agents or buffers that reduce the heat of reaction at the oxide-aluminum interface by way of saponification in order to promote a smoother, more uniform finish thickness.
When an electroactive modifier is added to the electrolyte, it is important that as it reacts with the anodic oxide, the reaction product between the modifier and the finish maintains conductivity within the central pore. This enables the ionic transport necessary to enable the finish to continue outward growth. The modifier chemistry should not degrade when exposed to the anodizing electrical process parameters. If it does, the resultant reaction products should not interfere with ion exchange within the pore. In contrast, although the chromate ion does carry a charge and reacts with the forming anodic oxide initially, the natural passive quality of chromium stabilizes the would-be ion exchange within the forming column wall and shuts down finish growth in Type I anodizing.
Development of a new electrolyte formulation
 Figure 9: TEM images of the composite anodic oxide finish. Note the appearance of the composite finish microstructure appears as a hybrid between a Type I and Type II finish.Theoretically, the extent to which the engineering properties of the anodic oxide finish are altered is reflected in how much modifier is incorporated into or reacted with the anodic oxide structure. Selection of an effective electrolyte modifier must take into consideration how the final structure of the anodic oxide finish will change as a result of its addition. A modifier that shuts down the ion pump (one that contains a passive counterion component) will yield a finish that is thin and compact. Whereas, one that maintains the conductivity of the column wall will enable the finish to grow thicker and exhibit a porous structure. A modifier that cannot withstand the electrical parameters of the subject process may not impact the structure of the finish at all.
Figure 9: TEM images of the composite anodic oxide finish. Note the appearance of the composite finish microstructure appears as a hybrid between a Type I and Type II finish.Theoretically, the extent to which the engineering properties of the anodic oxide finish are altered is reflected in how much modifier is incorporated into or reacted with the anodic oxide structure. Selection of an effective electrolyte modifier must take into consideration how the final structure of the anodic oxide finish will change as a result of its addition. A modifier that shuts down the ion pump (one that contains a passive counterion component) will yield a finish that is thin and compact. Whereas, one that maintains the conductivity of the column wall will enable the finish to grow thicker and exhibit a porous structure. A modifier that cannot withstand the electrical parameters of the subject process may not impact the structure of the finish at all.
Parkhutik et.al. first demonstrated how electroactive films of certain conjugated polymers could be deposited on anodized silicon in the early 1990s.8 In 1993, the author recognized similarities between the aluminum anodizing process and the oxidative polymerization reactions for certain conjugated polymers, as well as the electroactive characteristics of these polymers with protonic acid doping. These characteristics indicated anodizing and electrodeposition of the polymer could be carried out simultaneously, producing composite polymer-metal oxide finishes.9-11 The research goal surrounding this hypothesis was the formation of a transition layer that would enable chemical bonding between polymers and aluminum. A true polymer-metal transition layer would be metallurgically bound to the aluminum substrate by way of the anodic oxide and offer sites for polymer bonding at the oxide surface. Finishes such as these would have much utility in the automotive industry where delamination of polymer seals, gaskets, labels and paints constitute common failure modes.
By modifying the sulfuric acid electrolyte to include one of these polymers, uniform, continuous finishes have been electrochemically formed, which exhibit structural modification and polymer-phase inclusion. The composite anodic oxide finish exhibits a hybrid microstructure that integrates the columnar structure of the Type II and III finishes with the irregular, skewed appearance of the Type I finish. Correspondingly, the finish exhibits improved corrosion and wear resistance over conventional anodic oxide finishes. See Figure 9.
The anodizing process for the described polymer-anodic aluminum composite oxide was reduced to practice and developed for large-scale production. It was patented and is currently commercially available.12 To date, engineering application of the composite anodic oxide finish has shown increased corrosion and wear resistance, ability to be dyed and UV stability. In addition, the finish exhibits superior adhesion of subsequent polymer layers and functions as a transition layer for bonding.13-15 In an effort to understand the performance improvements, scientific characterization and testing of the composite finish has been performed to compare with standard anodic finishes in the laboratory and in various field applications. Several years in full-scale production have shown the process is easy to handle, efficient and less expensive to operate than conventional hard anodizing processes.
Scientific Characterization16
High intensity infrared (FT-IR) spectrographic analysis
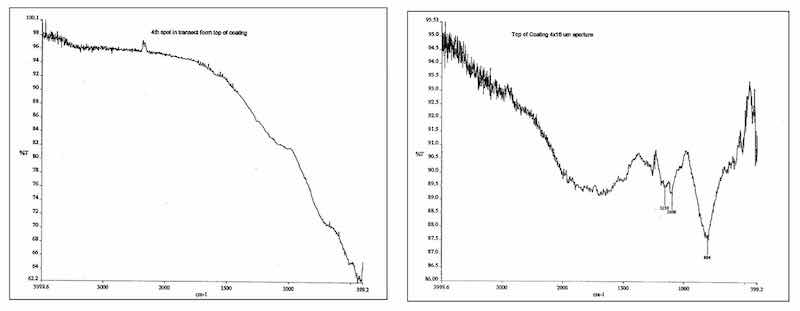 Figure 10: IR spectrum of composite finish adjacent to the aluminum substrate is identical to a conventionally anodized Type II film. The low-end absorbance is typical for inorganic species. IR spectrum of composite finish exhibits a shift in the inorganic spectrum toward the more chemically reactive species of sulfate and hydroxide. Organic absorbances are also present in the higher IR indicative of inclusion of the electroactive polymer additive as well as more hydration.Transmission Electron Microscope (TEM) sections of a Type II unsealed anodic film and composite films were analyzed by way of high intensity Fourier Transform Infrared Spectrographic analysis to determine if the change in structure could be corroborated with a change in composition due to the presence of the polymer modifier. The energy source for the instrument was hooked up to the synchrotron light source at Brookhaven National Laboratory in Brookhaven, New York. The resolution of the instrument was 4μ to 5μ. Sections of each coating were analyzed using a 4μ by 30μ aperture. Data was collected from the aluminum-anodic film interface (bottom), the film center and the top. The films measured 20μ to 25μ thick.
Figure 10: IR spectrum of composite finish adjacent to the aluminum substrate is identical to a conventionally anodized Type II film. The low-end absorbance is typical for inorganic species. IR spectrum of composite finish exhibits a shift in the inorganic spectrum toward the more chemically reactive species of sulfate and hydroxide. Organic absorbances are also present in the higher IR indicative of inclusion of the electroactive polymer additive as well as more hydration.Transmission Electron Microscope (TEM) sections of a Type II unsealed anodic film and composite films were analyzed by way of high intensity Fourier Transform Infrared Spectrographic analysis to determine if the change in structure could be corroborated with a change in composition due to the presence of the polymer modifier. The energy source for the instrument was hooked up to the synchrotron light source at Brookhaven National Laboratory in Brookhaven, New York. The resolution of the instrument was 4μ to 5μ. Sections of each coating were analyzed using a 4μ by 30μ aperture. Data was collected from the aluminum-anodic film interface (bottom), the film center and the top. The films measured 20μ to 25μ thick.
The trends in the infrared data collected from the bottom of the anodic oxide finishes to the top demonstrate a shift in the formation and amount of active hydroxide and sulfate groups. The Al-O feature at approximately 750 cm-1 dominates, but shifts upward in the spectra as the sulfate peaks at 1000cm-1 to 1100cm-1 become larger and more defined. With the development of the sulfate absorption, hydroxide absorption becomes pronounced. This actually makes sense, as the surface of the anodic film, regardless of type or formulation, should exhibit more hydration.
In order to identify the molecular species of the oxide, as formed during conventional anodization, a hot- water sealed Type III coating was processed within argon plasma. With increased exposure time within the plasma, a residue developed on the surface of the coating. Infrared analysis of the residue determined absorption characteristics for hydrated aluminum sulfate and/or aluminite: Al2(SO4) • 18H2O and/or Al2(SO4)(OH)4 • 7H2O.
The composite coating exhibited evidence of inclusion of the electroactive modifier with absorption in the higher infrared (IR) wave numbers. The spectral shifts toward the higher IR wave numbers were noted from the substrate, where the inorganic absorbances were most pronounced, to the middle and at the surface portions of the modified AAO where absorption of both OH-1 and carbon-based inorganic salts were detected within the composite film. See Figure 10.
These results indicate the presence of a compositional gradient from the substrate to the finish surface. This shows that the portions of the film that remain in contact with the electrolyte, in addition to hydration, will also adsorb active anion species from the electrolyte. Since the central pore is in constant contact with the electrolyte, a compositional and reactivity gradient across the porous structure also forms. The formation of these reactive sites provides the necessary link for the anodic oxide to be modified through additives to the electrolyte.
X-ray photoelectron spectroscopy
In order to determine how the polymer modifier had reacted with the anodic oxide structure during anodizing, X-ray Photoelectron Spectroscopy (XPS) studies were performed on Type II and on two groups of composite anodic oxide samples at the University of Linköping in Linköping, Sweden. The composite samples were differentiated by the time of exposure to the anodizing process. One group of samples was representative of the typical exposure time of 60 minutes and the other group was representative of an extended exposure time of 180 minutes. The samples were anodized and mechanically removed to sealed containers to prevent surface contamination through handling or by ambient air. This insured the analysis results would reflect the actual anodized surface composition from a depth of 0 to 30 Angstroms.
The Type II and composite samples anodized for the typical exposure time exhibited the presence of sulfur as sulfate. Additional oxygen and aluminum was also detected. The high-level binding energy component for oxygen corresponded to H2O and OH-, which supported the infrared data. The aluminum detected in the film surface of these samples was not metallic in nature. The low-binding energy component was typical for amorphous disordered aluminum oxide. The composite sample did show chemical inclusion of the electroactive modifier.
XPS analysis of composite samples anodized for an extended period of time (3 hours rather than 1 hour) determined strikingly different surface constituents. More of the electroactive additive was detected in the surface of these samples. No sulfur was determined. Evidence of metallic aluminum and copper (as Cu2O) was also identified. The results were important because they indicated these species were actually adsorbed from the electrolyte and not a function of the anodizing process.
The results of the XPS study determined the polymer molecule is altered as it oxidizes during the anodization reaction. Saturated and π-conjugated carbons were noted with distinct linkages to the disordered hydroxide structure as HO-C=O, O-C=O and C-OH. These ligands are unstable and indicate why conduction is maintained through the porous columnar structure, even though the long-chain molecule has reduced the oxide structure producing the somewhat skewed appearance documented through TEM. Non-protonated aluminum salts of the polymer, found to be present at the surface of the composite anodic oxide during IR analysis, show that through protonic conduction of the forming anodic oxide, aluminum ions also reacted with the electroactive polymer modifier.
Adhesion testing
 Figure 11: TEM image documenting the presence of fibrils bridging the interface between the composite anodic oxide finish and the rubber shell, which had been molded over the anodized aluminum component.Two hundred samples each of aluminum alloy 6061 components were finished with a hard version of the composite coating (Type III equivalent), dyed black and sealed two different ways. One set of one hundred components was sealed with hot water and one set of one hundred components was sealed in a duplex fashion, first with nickel acetate and followed by a sodium dichromate seal. The sample sets were subjected to four permutations of cleaning and treatment with a bonding agent: (1) no cleaning with no bonding agent; (2) cleaned with no bonding agent; (3) no cleaning with bonding agent; and, (4) cleaned with bonding agent. All were coated with adhesive and over-molded with a rubber-based form.
Figure 11: TEM image documenting the presence of fibrils bridging the interface between the composite anodic oxide finish and the rubber shell, which had been molded over the anodized aluminum component.Two hundred samples each of aluminum alloy 6061 components were finished with a hard version of the composite coating (Type III equivalent), dyed black and sealed two different ways. One set of one hundred components was sealed with hot water and one set of one hundred components was sealed in a duplex fashion, first with nickel acetate and followed by a sodium dichromate seal. The sample sets were subjected to four permutations of cleaning and treatment with a bonding agent: (1) no cleaning with no bonding agent; (2) cleaned with no bonding agent; (3) no cleaning with bonding agent; and, (4) cleaned with bonding agent. All were coated with adhesive and over-molded with a rubber-based form.
Adhesion testing of the samples was based in ASTM B571 “Test Methods for Adhesion of Metallic Coatings,” paragraph 11, and “Peel Test.” The rubber coating was cut and pulled at an angle of 90o to the surface of the piece with efforts to maintain the rate of pull. Following testing, the delaminated surfaces were evaluated to determine the point of failure. Inadequate adhesion was considered as failure in the coating-substrate interface.
Comparison of the various samples determined that there was little difference in adhesion between the samples processed with the hot-water seal and those with the duplex seal. In fact, it was found that the degreasing operation, performed prior to treatment with the adhesion promoter, could successfully be eliminated without compromising adhesion of the rubber form. Upon consideration of the adhesion testing results, use of the composite finish as a stand-alone finish with a hot-water seal, was selected. Adhesion of the bonding agent was as good to the composite finish as it was to the dichromate seal. Implementation of the composite finish, with the hot-water seal, eliminated the use of the heavy-metal seal and the degreasing process, promoting a more environmentally friendly finish and process. It also saved time and money.
Detailed TEM analysis of samples prepared for comparative adhesion testing disclosed the presence of fibrils bridging the interface between the composite anodic oxide finish, that had been hot-water sealed, and the organic bonding agent or adhesive. This feature was not present in the samples processed with the dichromate seal, demonstrating the absence of chemical bonding between the bonding agent and the dichromate coating. The darker appearance of the fibrils indicates that they consist of low-atomic number elements. EDS analysis determined carbon as a significant constitutive element, although the constituents of the anodic oxide were also detected. See Figure 11.
Conclusion
 Figure 12: Examples of applications in which the composite anodic oxide is used as a transition layer for bonding aluminum with a polymer. The application on the left is an automotive power- steering module. The application on the right is a fire hose nozzle.Effective bonding of dissimilar materials often incorporates a transition layer that comprises elements of the components to be joined. Therefore, if the bonding of a metal to an adhesive is to be considered, the transition layer should contain constituents that would bond and combine with the metal and constituents that would, in turn, bond and combine with the adhesive. The adhesive constituents would most likely be polymer in nature.
Figure 12: Examples of applications in which the composite anodic oxide is used as a transition layer for bonding aluminum with a polymer. The application on the left is an automotive power- steering module. The application on the right is a fire hose nozzle.Effective bonding of dissimilar materials often incorporates a transition layer that comprises elements of the components to be joined. Therefore, if the bonding of a metal to an adhesive is to be considered, the transition layer should contain constituents that would bond and combine with the metal and constituents that would, in turn, bond and combine with the adhesive. The adhesive constituents would most likely be polymer in nature.
The design of a metal-polymer transition layer has often been approached via mechanical or chemical roughening of the surface followed by chemical adhesion promoters, conversion coatings or other types of primers. A mechanical attachment to the metal is often insufficient, and interfacial breakdown by way of filiform corrosion is a frequent failure mode. The use of a primer layer improves the attachment, but they are often thin and subject to abrasion. Moreover, if they have no conversion effect on the surface of the metal, they will provide no barrier to corrosion breakdown at the interface.
Aluminum presents a unique characteristic in that it naturally forms a passive layer. The passive layer or passive oxide, although extremely thin (about 1 to10 nanometers), effectively protects the surface from intrusion of external anions. The bond to the metal is one of chemisorption and, although effective, is easily abraded. Nevertheless, the formation of a conversion coating that employs anions that will bond to the passive layer forms an effective, albeit fragile, transition layer for adhesive bonding to aluminum.
Anodizing builds on the characteristics of the passive layer: not only is the bond to the aluminum enhanced through the formation of the anodic oxide, the layer also is substantially thicker, and the structure is more abrasion resistant. AAO can be tailored to be as thin as 50 to 70 nanometers (flash anodizing) and as thick as any hard anodic oxide, in excess of 30 microns.
The porous structure of the anodic oxide also prohibits the intrusion of anions from the electrolyte, but offers ionic sites for external anion attachment. By introducing an electroactive polymer additive to the electrolyte, polymer ligands attach to the AAO structure, providing sites for actual chemical bonding of adhesives or other bonding media. Composite anodic oxides are currently in use in many industrial applications. See Figure 12.
Acknowledgements: Special thanks to Ms. Joy Kaufman for the schematic artwork and to Mr. Michael Pavia for reviewing the text.
References
- Brown, L., Jackman, J., “Aluminum Extrusion Aids Auto Lightweighting”, Design News, January, 2015.
- Murr, L., Interfacial Phenomena in Metals and Alloys, Addison-Wesley Publishing Co., 1975.
- Csokan, P., „Nucleation Mechanism in Oxide Formation During Anodic Oxidation of Aluminum“, Advances in Corrosion Science and Technology, M. G. Fontana, et.al., editors, 1980.
- Sato, N.; “Basics of Corrosion Chemistry”, Green Corrosion Chemistry and Engineering: Opportunities and Challenges, First Edition, ed. Sanjay K. Sharma, Wiley-VCH Verlag GmbH and Co. KGaA, 2012.
- Runge, J., “Advancements in Anodizing: Improved Finish Properties through Electrolyte Modification”, from the conference proceedings of Aluminium 2000, Florence, 2007.
- Runge, J., “Formation of Porous Anodic Oxide Finishes: A New Approach and Theory”, from the conference proceedings of Aluminium 2000, Florence, 2007.
- Stumm, W., Furrer, G., “The Dissolution of Oxides and Aluminum Silicates; Examples of Surface- Coordination-Controlled Kinetics”, Aquatic Surface Chemistry; Chemical Processes at the Particle-Water Interface, ed. Werner Stumm, Wiley & Sons, Zurich, 1989.
- Parkhutik, V., Martinez-Duart, J.,”Deposition of Electroactive Polymer Films onto Porous Silicon Layers”, Journal of the Electrochemical Society, 140, No.6, L94-L95, 1993.
- Johnson, B., Park, S., “Electrochemistry of Conductive Polymer XIX, Oxidation of Electroactive Monomers at Bare and Polymer – Modified Platinum Electrodes Studied by Electrochemical Impedance Spectroscopy, Journal of the Electrochemical Society, 143, No. 4: 1269-1276, 1996.
- Yue,J., Wang, Z., Cromack, K., Epstein, A., MacDiarmid, A., “Effect of Sulfonic Acid Group on Electroactive Polymer Backbone”, Journal of the American Chemical Society, 113, No. 7: 2665 – 2671, 1991.
- Huang, W., Humphrey, B., MacDiarmid, A., “A Novel Conducting Polymer – Morphology and Chemistry of Its Oxidation and Reduction in Aqueous Electrolyte”, Journal of the Chemical Society, Faraday Transactions I, 82: 2385 – 2400, 1986.
- US patent no. 5,980,723
- Runge, J., “Toughness: The Key to Improved Anodic Oxide Finish Performance”, published in the proceedings for the annual technical conference for the Aluminum Anodizers Council, 2006.
- Runge, J. Pomis, A., Nussbaum, T., “Insights Regarding the Adhesion Mechanism for Supplementary Organic Coatings on Porous Anodic Films”, published in the proceedings of the Aluminium Essen conference, 2002.
- Runge, J., Pomis, A., “Continued Development in Chrome-Free Anodic Oxide Finishes for Aluminum: Evaluation of Selected Mechanical Properties”, published in the proceedings of the American Electroplaters and Surface Finishers Society Aerospace/Aircraft Forum August 27-29, 2002.
- Runge, J., Pomis, A., “Anodic Oxide Film Formation: Relating Mechanism to Composition and Structure”, Proceedings of the AESF SUR/FIN 2000 Technical Conference, AESF, June 2000.
 The Metallurgy of Anodizing Aluminum" is available at https://link.springer.com/book/10.1007/978-3-319-72177-4 Dr. Jude Mary (Judy) Runge’s career as a metallurgical engineer and surface finishing expert spans almost 40 years in industrial, government and academic professional settings. Beginning in 1982 at Northrop Corporation, Defense Systems Division, and culminating today as a Principal Engineer, Surface Finishing at Apple (since 2019), she is recognized internationally as a nonferrous specialist focusing on materials engineering problem solving that utilizes her expertise as a surface scientist and manufacturing process engineer, providing characterization for product development, failure analysis and metallurgical support to the aluminum finishing industry. She is well known for her work in anodizing that led to a new theoretical treatment for porous oxide formation. Dr. Runge received her Ph.D. in metallurgy at the University of Illinois at Chicago under Dr. Michael McNallan. A tireless educator, Dr. Runge has authored numerous papers and given seminars worldwide; she is the Education Chair for the Aluminum Anodizers Council since 2008. Her book, “The Metallurgy of Anodizing Aluminum”, published by Springer Nature in 2018, is one of her biggest personal achievements. Judy is third of nine children and the first in her family to attend college/university. She is the mother of 4 and grandmother of 8. She believes her success is the result of great personal grit and passion for science, which enabled her hard work and very often, hard decisions. She owes a great deal of her career to her mother, who continuously challenged and supported her. She is grateful to her husband, Thomas Nussbaum, for his love and support and for admitting always how proud he is of her.
The Metallurgy of Anodizing Aluminum" is available at https://link.springer.com/book/10.1007/978-3-319-72177-4 Dr. Jude Mary (Judy) Runge’s career as a metallurgical engineer and surface finishing expert spans almost 40 years in industrial, government and academic professional settings. Beginning in 1982 at Northrop Corporation, Defense Systems Division, and culminating today as a Principal Engineer, Surface Finishing at Apple (since 2019), she is recognized internationally as a nonferrous specialist focusing on materials engineering problem solving that utilizes her expertise as a surface scientist and manufacturing process engineer, providing characterization for product development, failure analysis and metallurgical support to the aluminum finishing industry. She is well known for her work in anodizing that led to a new theoretical treatment for porous oxide formation. Dr. Runge received her Ph.D. in metallurgy at the University of Illinois at Chicago under Dr. Michael McNallan. A tireless educator, Dr. Runge has authored numerous papers and given seminars worldwide; she is the Education Chair for the Aluminum Anodizers Council since 2008. Her book, “The Metallurgy of Anodizing Aluminum”, published by Springer Nature in 2018, is one of her biggest personal achievements. Judy is third of nine children and the first in her family to attend college/university. She is the mother of 4 and grandmother of 8. She believes her success is the result of great personal grit and passion for science, which enabled her hard work and very often, hard decisions. She owes a great deal of her career to her mother, who continuously challenged and supported her. She is grateful to her husband, Thomas Nussbaum, for his love and support and for admitting always how proud he is of her.































